Applying Fluoroquinolone Pharmacokinetics, Pharmacodynamics, and Updated Clinical Breakpoints for Gram-Negative Pathogens to Determine Optimal Dosing
1/30/2019
Written by: Eric Wenzler, University of Illinois at Chicago, Chicago, IL
Antimicrobial-resistant bacteria are responsible for significant morbidity, mortality, and excess healthcare costs, and their prevalence is increasing at a rate faster than the antimicrobial development pipeline.1 FDA-approved antimicrobial doses represent estimates across a patient population in order to maximize generalizability and probability of clinical effect as it would apply to “most” patients. Unfortunately, the increase in bacterial resistance and medical complexity of our patients means that these population estimates no longer apply to “most” patients, or at least not to the patients in whom optimizing antimicrobial doses matters the most. Fortunately, the principles of antimicrobial pharmacokinetics (PK) and pharmacodynamics (PD) can help optimize dosing regimens and tailor therapy for each patient individually.
In the January 2018 CLSI AST News Update, the basic principles behind PK and PD were presented. This article expands on that knowledge, demonstrating how PK/PD principles are applied in practice by clinicians when managing patients with challenging infections.
Pharmacokinetic Principles of Fluoroquinolones
In the previous CLSI AST News Update, PK was summarized as the evolution of a drug’s concentration, throughout the body and over time, after a dose of drug is administered. PK is governed by 4 principles: absorption, distribution, metabolism, and elimination, which are evaluated when deliberating the optimum treatment regimen for a patient. Absorption applies primarily to oral antimicrobials, as there is no absorption phase for antimicrobials administered intravenously (modern antimicrobials are rarely administered by topical or intramuscular routes). For oral antimicrobials, absorption is most commonly discussed in the context of bioavailability (F or fraction absorbed, expressed as %). Absolute bioavailability is the total amount of drug in circulation after oral administration compared to that after IV administration, whereas relative bioavailability is one oral formulation compared to another. Bioavailability incorporates both the rate and extent of absorption of the drug and can be affected by many factors including: gastrointestinal transit time, drug disintegration and dissolution, the activity of physiological transporters (ie, molecules in the human body that transport molecules to the cytoplasm of cells, for example in the kidney), interactions with other drugs, and food. Among the commonly used fluoroquinolones, levofloxacin and moxifloxacin have virtually 100% bioavailability meaning that the route of administration can be converted between IV and PO in a 1:1 ratio with no adjustment.2,3 The bioavailability of ciprofloxacin and delafloxacin are 70% and 59% respectively, meaning that a slightly higher dose must be given orally in order to match the exposure achieved after an IV dose.4,5
Distribution is most often referred to in the context of the volume of distribution of a drug (Vd). This is most correctly termed the apparent Vd, as Vd is not an actual biological or physiologic parameter. Vd is the volume that would be necessary to account for the total amount of drug in the body, if that drug were present in the entire body at the same concentration as in the plasma relative to the dose administered. Simply put, apparent Vd is merely a conversion factor required to convert a dose (expressed as mg or g) into a concentration (expressed as µg/mL or mg/L), as it is measured in the body. Vd is commonly normalized to body weight and expressed as L/kg. For example, if a 1000mg dose of vancomycin is administered intravenously and then the concentration measured in the patient’s blood 1 hour later is 17mg/L, the apparent volume that that dose must have been dissolved in is 58.9 L (1000mg/17mg/L=58.9L, or 0.7L/kg for 80kg patient). A volume of distribution ≥0.7L/kg is typically considered large and is associated with tissue concentrations exceeding those in plasma and adequate penetration into extravascular body sites. The fluoroquinolones have Vd values ranging from 0.6 to ≥2L/kg and are widely distributed throughout the body into lung, skin, fat, muscle, cartilage, bone, and many bodily secretions. The distribution of a drug is affected by its intrinsic properties such as its molecular size, charge, hydrophilicity or lipophilicity, and protein binding.
Metabolism occurs primarily in the liver and intestines. The vast majority of antimicrobials (including the fluoroquinolones) are not extensively metabolized, and for those that are, formed metabolites are rarely microbiologically active and thus do not contribute to overall drug efficacy.
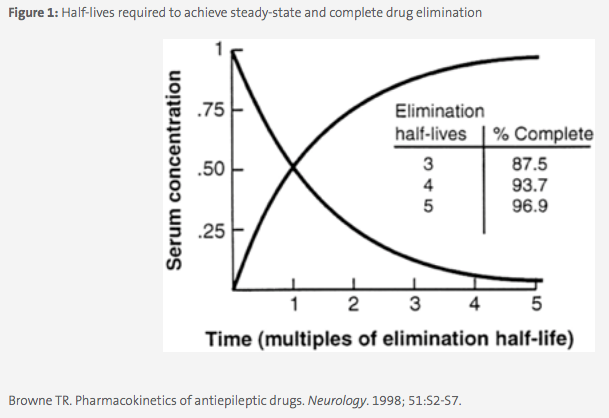
Elimination is the most important PK parameter for antimicrobials and is discussed in terms of total body clearance (CLT ) as the volume of blood removed of drug per unit time (eg; L/h). Almost all antimicrobials are primarily cleared through the kidneys and this is affected by the size of the drug, its protein binding, and the health of the patient’s kidneys. Clearance is used in part to determine the elimination half-life of the drug, which is the time it takes for the drug concentration in the body to be halved. Half-life is a more clinically useful parameter as steady-state, or the state in which drug concentration is in equilibrium in the body, occurs mathematically after 5 half-lives (eg, 50%, 75%, 87.5%, 93.8%, 96.9%, see Figure 1). The frequency at which a drug is dosed is therefore based in part on the half-life, and a typical rule of thumb is to dose a drug after at least 3 half-lives, or once ~87.5% of the dose has been eliminated from the body. For example, the half-life of levofloxacin in plasma is approximately 7.5 hours, therefore the drug is dosed clinically every 24 hours.2 Estimates of the patient’s clearance and subsequent drug half-life are used to make dosing adjustments for patients with increased or decreased renal function, including patients on dialysis.
Application of Fluoroquinolone PK/PD Principles: Case Examples
A 54-year-old Caucasian female patient is admitted to your institution from her skilled nursing facility and diagnosed with bacteremia due to Pseudomonas aeruginosa. The microbiology laboratory reports a MIC of 2µg/mL to levofloxacin for this isolate, which was within the susceptible range, according to the M100 2018 edition.6 The patient’s estimated creatinine clearance (CrCl, a measure of kidney function) is normal at 80mL/min. The team would like to de-escalate from intravenous piperacillin-tazobactam and send her back to her facility on oral levofloxacin. Can oral levofloxacin be used to treat this infection? If so, what dose should be used? To answer this question, we first need to know the PK/PD index of efficacy for levofloxacin, which is the AUC/MIC ratio. Next, we need to know the AUC/MIC target for Gram-negative pathogens. AUC/MIC targets are initially established by in vitro or in vivo dose fractionation studies and (ideally) validated in human clinical studies. We can refer to a clinical study in patients with hospitalacquired bacterial pneumonia, which demonstrated higher rates of pathogen eradication when a levofloxacin AUC/MIC of at least 87 was achieved.7 Therefore, we know we need an AUC of at least 174 mg x h/L for this patient, as the MIC is 2 µg/mL. We can next approach this question in two ways. First, the levofloxacin package insert is referenced to see what AUC is achievable with oral dosing. The maximum oral dose (750mg every 24 h) provides an AUC of 101±20 mg x h/L in healthy volunteers.2 Based on this initial check, it does not seem as if oral levofloxacin would be an appropriate recommendation for this patient. However, more accurate calculations can be used to answer this question definitively. To do this, the patient’s specific total body clearance (CLT ) of levofloxacin is evaluated. This can be achieved by using an equation relating patient-specific CrCl to CLT of levofloxacin from a population PK study from the published literature, and then determining the AUC via the equation AUC = Dose/CL. Ideally, the population PK analysis would have been performed in a population that matches or closely resembles the patient in question. In this example we will utilize the equation from a group of patients with serious community-acquired infections.8 The CL equation is:
- 5.945+race+(agex-0.032)+(CrClx0.07)
- To solve for our patient: CL=5.945–1.486+(54x-0.032)+(80x0.07)=8.33L/h
- So an AUC of 174 = dose/8.33. Solving for dose we get 1449.4 mg, which is almost twofold the maximum daily dose of 750 mg.
Therefore, despite the fact that the MIC in this case is considered susceptible, levofloxacin given PO would not be an appropriate recommendation for this patient. Because fluoroquinolone oral bioavailability is the same as IV, IV levofloxacin is also not an option. This discordance between fluoroquinolone breakpoints and achievable systemic PK has been recognized and reported for years.9 This discordance is one reason the fluoroquinolone susceptibility breakpoints are now lower in CLSI M100-Ed29.10 The impetus for this change stems from several factors including the increase in MICs to fluoroquinolones among Gram-negative pathogens, advances in PK/PD knowledge and technology, and the paucity of clinical data supporting the original breakpoints.11
The impact of this change can be examined in part through evaluating the PK of ciprofloxacin and revised clinical breakpoints against Enterobacteriaceae. From the ciprofloxacin package insert, the AUCs after a 500mg and 750mg every 12-hour oral dose, respectively, are 27.4 and 31.6mgxh/L. To achieve the aforementioned AUC/MIC target of 87, these two doses could be used effectively for MIC values up to 0.25µg/mL (the updated susceptibility breakpoint for ciprofloxacin against Enterobacteriaceae approved for CLSI M100-Ed29). Under the old breakpoints, both 0.5 and 1µg/mL would have been considered susceptible but would not be achievable from a PK/PD perspective in a patient with normal renal function. In addition to correlating with PK/PD targets, the revised breakpoints are closer to those published by EUCAST.12 CLSI has submitted a rationale document outlining the data supporting the change to the US FDA for their review and potential recognition. In anticipation of the delay until FDA recognition of the revised breakpoints, the performance of fluoroquinolone disk diffusion and Etest has been compared to reference BMD and demonstrated adequate performance using the updated 2019 CLSI breakpoints.11
To illustrate further, we can examine a case of a patient with pneumonia due to Klebsiella pneumoniae. The patient is a 72-year-old female with a CrCl of 45mL/min and an MIC to ciprofloxacin of 0.25µg/mL. Given her age and slightly impaired renal function, we can assume her patient-specific AUC after either a 500mg or 750mg every 12 hour dose would be even higher than those listed in the package insert for patients with normal renal function, and therefore either dose would be appropriate to achieve the needed AUC of 21.75mgxh/L. To be sure we can apply a population PK equation relating CrCl to CLT derived from 44 elderly patients with lower respiratory tract infections:13
- CLT=8+0.21xCrCl
- Therefore, CLT=8L/h+0.21x45=17.5L/h.
- AUC=Dose/CL; 21.75=Dose/17.5; 380.63mg.
These estimations suggest that the 500 mg every 12-hour dose would be adequate for this patient and that the 750 mg dose can be avoided, which may reduce potential dose-related toxicities in this elderly patient.
Summary
While these practical examples provide patient-specific dosing recommendations and allow us to assess the appropriateness of an antimicrobial dosing regimen beyond simply referring to the pathogen’s MIC, more advanced applications of these principles that incorporate infection site and variability in both MIC and PK measurements are available.14 The probability of optimal clinical outcomes is maximized when antimicrobial PK/PD targets are achieved. Due to the rise in bacterial resistance and medical complexity of patients, standard population-based doses of these agents rarely meet PK/PD targets in the patients who are most dependent on it for survival, such as those who are critically ill and have pathophysiological derangements affecting their PK. As such, it is necessary for clinicians to understand and employ precision medicine via PK/PD principles when determining antimicrobial dosing regimens.
References
1 Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States, 2013. http://www.cdc.gov/drugresistance/threat-report-2013/pdf/ar-threats-2013-508.pdf. Accessed January 26, 2015.
2 LEVAQUIN (levofloxacin) [package insert]. Titusville, NJ; Janssen Pharmaceuticals, Inc. Revised September 2008. https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/020634s065,020635s071,021721s032lbl.pdf. Accessed March 20, 2017.
3 AVELOX (moxifloxacin) [package insert]. Whippany, NJ; Bayer Healthcare Pharmaceuticals, Inc. Revised July 2016. https://www.merck.com/product/usa/pi_circulars/a/avelox/avelox_pi.pdf. Accessed March 20, 2017.
4 CIPRO (ciprofloxacin) [package insert]. Whippany, NJ; Bayer Healthcare Pharmaceuticals, Inc. Revised July 2016. https://www.accessdata.fda.gov/drugsatfda_docs/label/2016/019537s086lbl.pdf. Accessed March 20, 2017.
5 BAXDELA (delafloxacin) [package insert]. Lincolnshire, IL; Melinta Therapeutics, Inc. Revised June 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208610s000,208611s000lbl.pdf. Accessed January 7, 2019.
6 CLSI. Performance Standards for Antimicrobial Susceptibility Testing. 28th ed. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute; 2018.
7 Drusano GL, Preston SL, Fowler C, Corrado M, Weisinger B, Kahn J. Relationship between fluoroquinolone area under the curve: minimum inhibitory concentration ratio and the probability of eradication of the infecting pathogen, in patients with nosocomial pneumonia. J Infect Dis. 2004;189(9):1590-1597.
8 Preston SL, Drusano GL, Berman AL, et al. Levofloxacin population pharmacokinetics and creation of a demographic model for prediction of individual drug clearance in patients with serious community-acquired infection. Antimicrob Agents Chemother. 1998;42(5):1098-1104.
9 Haeseker M, Stolk L, Nieman F, et al. The ciprofloxacin target AUC: MIC ratio is not reached in hospitalized patients with the recommended dosing regimens. J Clin Pharmacol. 2013;75(1):180-185.
10 CLSI. Performance Standards for Antimicrobial Susceptibility Testing. 29th ed. CLSI supplement M100. Wayne, PA: Clinical and Laboratory Standards Institute; 2019.
11 Humphries RM, Hindler JA, Shaffer K, Campeau SA. Evaluation of ciprofloxacin and levofloxacin disk and Etest using the 2019 Enterobacteriaceae CLSI breakpoints. J Clin Microbiol. 2018; 56:01797-18.
12 European Committee on Antimicrobial Susceptibility Testing. Breakpoint tables for interpretations of MICs and zone diameters, version 6. http://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Breakpoint_tables/v_6.0_Breakpoint_table.pdf. Accessed December 23, 2015.
13 Cios A, Wyska E, Szymura-Oleksiak J, Grodzicki T. Population pharmacokinetic analysis of ciprofloxacin in the elderly patients with lower respiratory tract infections. Exp Gerontol. 2014;57:107-113.
14 Bulik CC, Bader JC, Zhang L, et al. PK-PD Compass: bringing infectious diseases pharmacometrics to the patient’s bedside. J of Pharmacokinetics and Pharmacodynamics. 2017;44(2):161-177.
This article came from AST News Update, Volume 4, Issue 1 – January 2019 which is produced by the CLSI Outreach Working Group (ORWG). The ORWG is part of the CLSI Subcommittee on Antimicrobial Susceptibility Testing (AST) and was established in 2015. The formation of the working group originated in a desire to efficiently convey information regarding contemporary AST practices, recommendations, and resources to the clinical microbiology community. They welcome suggestions from you about any aspect of CLSI documents, educational materials, or their Newsletters.